Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder. According to recent projections, it will affect more than 1 million people in the US alone by 20301. PD progressively alters brain function to induce resting tremor, bradykinesia, postural instability, and rigidity, which arise due to an initially selective loss of nigrostriatal dopaminergic neurons followed by more widespread neurodegeneration, including in the cerebral cortex2. Though historically considered an idiopathic (‘sporadic’) disorder, at least 5–10% of PD cases are caused by mutations in certain genes, of which α-synuclein (SNCA) and leucine-rich repeat kinase 2 (LRRK2) are two of the most prominent hits in genome-wide association studies (GWAS)3. Hyperphosphorylated, aberrantly-folded, and aggregated forms of αSyn – together with altered organelles and lipid membranes – comprise Lewy bodies, the diagnostic neuronal inclusions of both sporadic and familial PD.
Methods
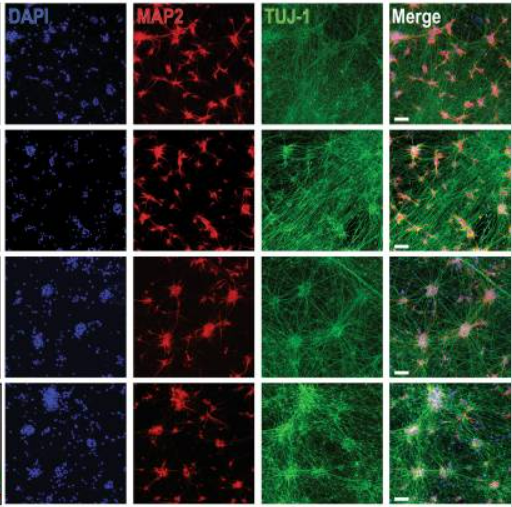
Evidence of cellular tetramers with a size and conformation distinct from the monomer was provided by (1) native gel electrophoresis; (2) intact-cell crosslinking; (3) STEM (scanning transmission electron microscopy); and (4) SE-AUC (sedimentation equilibrium analytical ultracentrifugation), which predicted a mass of tetramers purified from fresh human erythrocytes of 57,753 Da (4 monomers = 57,760).
Results
The LRRK2 G2019S mutation destabilizes the physiological tetramer:monomer equilibrium of αSyn and increases phosphorylation at pSer129, a marker of PD.
Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9481630/