Objective
Identification of genetic risk factors for Parkinson disease (PD) has to date been primarily limited to the study of single nucleotide variants, which only represent a small fraction of the genetic variation in the human genome. Consequently, causal variants for most PD risk are not known. Here we focused on structural variants (SVs), which represent a major source of genetic variation in the human genome. We aimed to discover SVs associated with PD risk by performing the first large-scale characterization of SVs in PD.
Methods
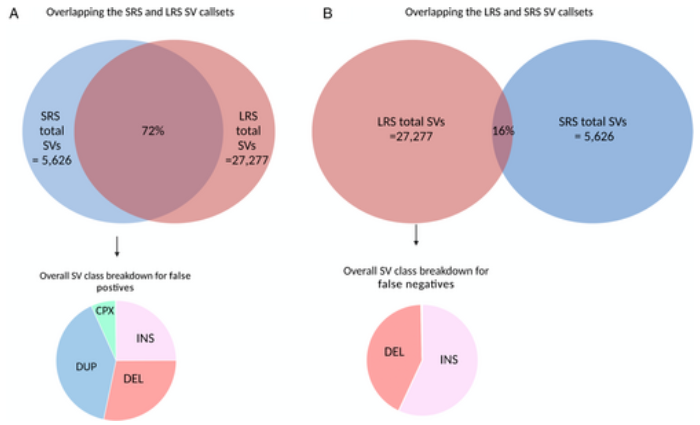
We leveraged a recently developed computational pipeline to detect and genotype SVs from 7,772 Illumina short-read whole genome sequencing samples. Using this set of SV variants, we performed a genome-wide association study using 2,585 cases and 2,779 controls and identified SVs associated with PD risk. Furthermore, to validate the presence of these variants, we generated a subset of matched whole-genome long-read sequencing data.
Results
We genotyped and tested 3,154 common SVs, representing over 412 million nucleotides of previously uncatalogued genetic variation. Using long-read sequencing data, we validated the presence of three novel deletion SVs that are associated with risk of PD from our initial association analysis, including a 2 kb intronic deletion within the gene LRRN4.
Interpretation
We identified three SVs associated with genetic risk of PD. This study represents the most comprehensive assessment of the contribution of SVs to the genetic risk of PD to date. ANN NEUROL 2023.
View Full Article: https://onlinelibrary.wiley.com/doi/full/10.1002/ana.26608