Abstract
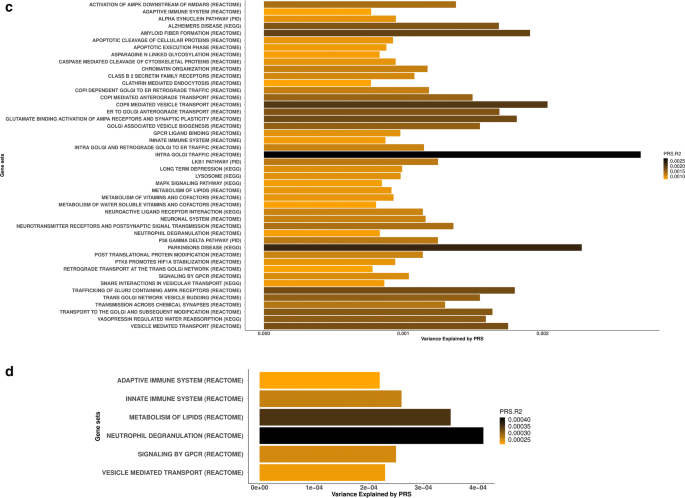
Polygenic inheritance plays a central role in Parkinson disease (PD). A priority in elucidating PD etiology lies in defining the biological basis of genetic risk. Unraveling how risk leads to disruption will yield disease-modifying therapeutic targets that may be effective. Here, we utilized a high-throughput and hypothesis-free approach to determine biological processes underlying PD using the largest currently available cohorts of genetic and gene expression data from International Parkinson’s Disease Genetics Consortium (IPDGC) and the Accelerating Medicines Partnership-Parkinson’s disease initiative (AMP-PD), among other sources. We applied large-scale gene-set specific polygenic risk score (PRS) analyses to assess the role of common variation on PD risk focusing on publicly annotated gene sets representative of curated pathways. We nominated specific molecular sub-processes underlying protein misfolding and aggregation, post-translational protein modification, immune response, membrane and intracellular trafficking, lipid and vitamin metabolism, synaptic transmission, endosomal–lysosomal dysfunction, chromatin remodeling and apoptosis mediated by caspases among the main contributors to PD etiology. We assessed the impact of rare variation on PD risk in an independent cohort of whole-genome sequencing data and found evidence for a burden of rare damaging alleles in a range of processes, including neuronal transmission-related pathways and immune response. We explored enrichment linked to expression cell specificity patterns using single-cell gene expression data and demonstrated a significant risk pattern for dopaminergic neurons, serotonergic neurons, hypothalamic GABAergic neurons, and neural progenitors. Subsequently, we created a novel way of building de novo pathways by constructing a network expression community map using transcriptomic data derived from the blood of PD patients, which revealed functional enrichment in inflammatory signaling pathways, cell death machinery related processes, and dysregulation of mitochondrial homeostasis. Our analyses highlight several specific promising pathways and genes for functional prioritization and provide a cellular context in which such work should be done.
Source: https://link.springer.com/article/10.1007/s00401-020-02181-3